Bio-informatica
Combinatie van informatica, statistiek en wiskunde om biologische onderzoeksvragen te beantwoorden

Over ons
Ons bio-informaticateam maakt deel uit van de Deskundigen-groep voor interdisciplinaire biowetenschappen (BIO).
Ons onderzoek is gespecialiseerd in omics-gegevensanalyse, algoritme- & pijplijnontwikkeling, data mining & multi-omics-gegevensintegratie met de focus op het opzetten van solide verwerkingsmethoden en tools, om een beter inzicht te bieden in proteomica, (meta-)genomica en de integratie met andere "omics".
We bieden ook bio-informaticaoplossingen voor een brede waaier aan biologische gegevens, zoals micro-organismen, menselijke cellen, muizen en planten, afkomstig van microbiologie, radiobiologie, biosfeer impactstudies laboratoria en kankeronderzoek op SCK CEN.
Recente publicaties
Als je meer wil weten over jouw gegevens of als je de meest optimale bio-informaticaoplossing wil vinden, staan wij open voor samenwerking. Om ons onderzoekswerk beter te leren kennen kan je onze recente publicaties raadplegen.
Expertise van het SCK CEN in omics-technologie
-

Genomica
- Volledige-genoomsequencing
- Metagenomica
- Amplicon sequencing
-

Epigenomica
- Globale genoommethylering (reeks, sequencing)
- Gerichte methylering (reeks, sequencing)
- ChiP-Seq
-

Transcriptomica
- Bulk RNAseq
- Micro RNAseq
- Lnc RNAseq
- Single cell RNAseq
-

Proteomica
- Massaspectrometrie
Gratis & open source tools
The tools below developed at SCK CEN are free, open source and have open access publications. These tools allow further support and contribution in the future research work of the scientific community.
De onderstaande instrumenten zijn gratis en open source, en werden ontwikkeld door het SCK CEN. De publicaties geven uitleg bij en delen het onderzoek van het SCK CEN met de hele onderzoeksgemeenschap.
Overzicht van de tools van SCK CEN
-
GenDisCal is bedoeld om grote aantallen bacteriële genomen systematisch met elkaar te vergelijken. Dit kan worden gebruikt om onbekende genomen te clusteren in groepen die tot dezelfde soort behoren, of als een manier om snel de soort te identificeren waartoe individuele stammen behoren. GenDisCal is in de eerste plaats ontworpen voor het gebruik van korte (3~8bp) oligonucleotide-frequenties, maar kan ook worden ingezet voor het berekenen van gemiddelde nucleotide-identiteit met NCBI-Blast (moet apart worden geïnstalleerd) of een benadering daarvan. GenDisCal heeft ook een grafische gebruikersinterface, waardoor het gemakkelijk te gebruiken is op zowel Windows als Linux.
-
MAGISTA is een uitlijningsvrije benadering om de volledigheid en zuiverheid van metagenomische bins te schatten. Meestal gebeurt dit met markergenen, maar slechts enkele van deze genen zijn universeel geconserveerd, hetgeen de nauwkeurigheid van dergelijke methoden beperkt voor nieuwe soorten die met behulp van metagenomica worden gevonden. Door oligonucleotide samenstellingen van 1kb- tot 50kb-fragmenten te gebruiken, maakt MAGISTA gebruik van intragenoomlimieten op interfragmentafstanden die consistent blijven voor de meeste bacteriële soorten. Door systematisch de verdelingen van deze interfragmentafstanden te analyseren met behulp van een random forest trainingsmodel dat realistische bins gebruikt, kan MAGISTA volledigheid en zuiverheid beter voorspellen dan op marker-genen gebaseerde methoden die berusten op universeel geconserveerde genen, hoewel het nog verre van nauwkeurig is.
-
BacPipe is een uitgebreide, snelle en rekenkundig eenvoudige bio-informaticapijplijn voor de analyse van sequenties van het volledige genoom van bacteriën, verkregen met sequencing-technologieën van de tweede en derde generatie. Gebruikers kunnen ervoor kiezen om ruwe sequencing-lezingen of contigs of scaffolds rechtstreeks in BacPipe te analyseren. De pijplijn is een geheel van geavanceerde, open toegankelijke bio-informaticatools voor kwaliteitsverificatie, genoomassemblage en -annotatie, en identificatie van het bacteriële genotype (MLST- en emm-typering), resistentiegenen, plasmide(n), virulentiegenen en single nucleotide polymorfismen (SNP's). De uitbraakmodule in BacPipe kan, samen met de SNP's en de metagegevens van de patiënt, worden gebruikt om gelijktijdig een groot aantal stammen te analyseren, om de evolutionaire relaties te begrijpen en snel bacteriële transmissieroutes te construeren. Belangrijk is dat BacPipe is ontworpen om meerdere instrumenten tegelijk te gebruiken, wat de tijd tot het verkrijgen van een resultaat aanzienlijk verkort. BacPipe biedt een geautomatiseerde pijplijn die een van de voornaamste hindernissen zal helpen overwinnen waarmee microbiologen worden geconfronteerd bij het analyseren en interpreteren van WGS-gegevens, en die de rechtstreekse toepassing ervan voor routinematige patiëntenzorg in ziekenhuizen en bij toezicht op de volksgezondheid en infectiebestrijding zal vergemakkelijken.
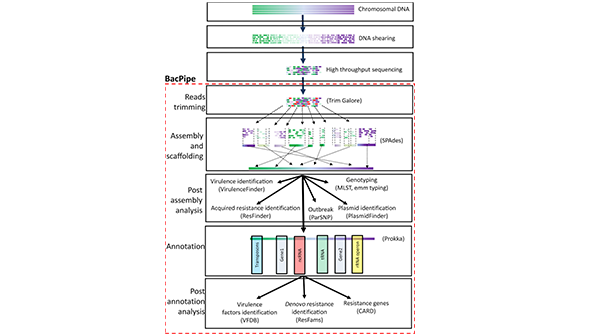
-
PCR-amplificatie van deze markergenen (16S, 18S, ITS, enz.) levert een aanzienlijke hoeveelheid kunstmatige sequenties op, die vaak chimaeren worden genoemd. CATCh combineert twee machine learning classifiers (referentiegebaseerd en de novo) die zijn ontwikkeld door de output van bestaande chimaeradetectietools te integreren in een nieuwe, krachtigere methode. Bij vergelijking van deze classifiers met bestaande tools in de reference-based of de novo modus werd een betere prestatie van deze ensemblemethode waargenomen op een brede waaier van sequencing-gegevens, waaronder gesimuleerde, 454 pyrosequencing, en Illumina MiSeq datasets. Aangezien dit algoritme de voordelen van verschillende individuele chimaeradetectie-instrumenten combineert, levert het robuustere resultaten op wanneer het wordt afgewogen tegen chimere sequenties met een lage stamdivergentie, een korte lengte van het chimere bereik, en verschillende aantallen stammen. Bovendien kon worden aangetoond dat de integratie van CATCh in de voorbewerkingspijplijn een gunstig effect heeft op de kwaliteit van de clustering in operationele taxonomische eenheden.
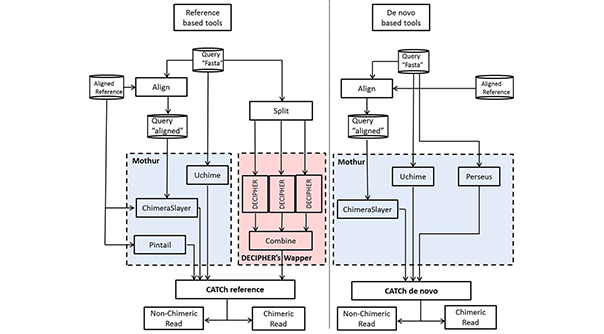
-
Voor 16S rRNA metagenomicastudies was de 454 pyrosequencing-technologie een van de meest gebruikte platforms, maar sequencing-fouten leiden nog steeds tot belangrijke problemen bij de gegevensanalyse (b.v. bij de clustering in taxonomische eenheden en de schatting van de biodiversiteit). NoDe (Noise Detector) is een algoritme voor foutcorrectie, ontworpen voor 454 pyrosequencing-gegevens. Het wordt getraind om die posities in 454 sequencing-lezingen te identificeren die waarschijnlijk een fout bevatten, en clustert vervolgens die foutgevoelige lezingen met correcte lezingen, wat resulteert in foutvrije representatieve lezingen. Het positieve effect van NoDe in 16S rRNA-onderzoek werd bevestigd door het gunstige effect op de precisie van de clustering van pyrosequencing-lezingen in operationele taxonomische eenheden. NoDe bleek een rekenkundig efficiënt denoising-algoritme te zijn voor pyrosequencing-lezingen, dat de laagste foutenpercentages opleverde in een uitgebreide benchmarking studie met andere denoising-algoritmen.
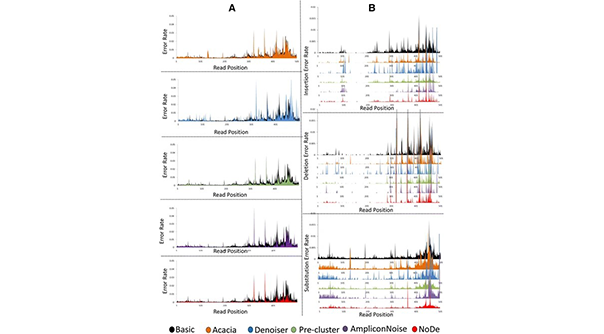
-
IPED is een krachtige denoising-tool voor het corrigeren van sequencing-fouten in Illumina MiSeq amplicon sequencing-gegevens. Het omvat een machine-leermethode die potentieel foutieve posities in sequencing-lezingen voorspelt op basis van een combinatie van kwaliteitsmaatstaven. Vervolgens wordt deze informatie gebruikt om die lezingen met een fout in te groeperen met correcte lezingen, wat resulteert in foutloze consensuslezingen. Dit wordt bereikt door mogelijk foutieve posities tijdens deze clusteringstap te maskeren. Het verlagen van de foutenmarge had een positief effect op de clustering van de lezingen in operationele taxonomische eenheden, met een bijna perfecte overeenkomst tussen het aantal clusters en het theoretische aantal soorten dat in de schijngemeenschappen aanwezig is.
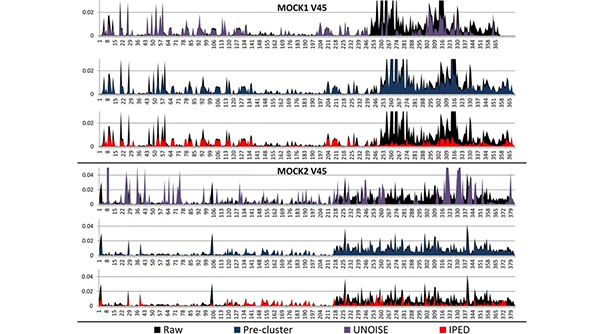
-
DynamiC is een taxonomisch gedreven regiospecifiek clusteringalgoritme. Het vervangt het gebruik van een standaard cut-off (gewoonlijk 97%) voor de clustering van 16S amplicons in operationele taxonomische eenheden (OTU's). Het gebruik van een algemene cut-off is twijfelachtig wanneer deze wordt toegepast op korte amplicons die slechts één of twee variabele regio's van het 16S rRNA-gen bestrijken. Dit leidt tot vertekeningen in de diversiteitscijfers en maakt het moeilijk om resultaten te vergelijken die zijn verkregen met amplicons welke voortkomen uit verschillende primersets. DynamiC daarentegen houdt rekening met de verschillende evolutiesnelheden van taxonomische lijnen om een dynamische en taxonomisch-afhankelijke OTU-clustering cut-off score te definiëren. Dit wordt geïmplementeerd door bij het bepalen van deze cut-off rekening te houden met de geamplificeerde variabele regio's en de taxonomische familie; een dergelijke drempel zal leiden tot meer robuuste resultaten en een nauwere overeenkomst tussen OTU's en soorten.
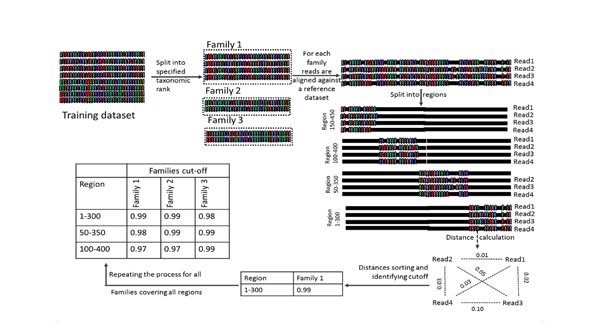
-
OCToPUS is een uitgebreide pijplijn die een optimale combinatie maakt van verschillende algoritmen, waaronder mothur: voor uitlijning en behandeling van lezingen, IPED: voor denoising, CATCh: voor chimaeradetectie, UPARSE: voor OTU-clustering en SPAdes voor pre-assembly filtering. OCToPUS behaalt het laagste foutenpercentage, het kleinste aantal spurious OTU's en de nauwste overeenkomst met de bestaande gemeenschap, terwijl het grootste aantal lezing behouden blijft in vergelijking met andere pijplijnen (in de vergelijkende analyse). De nieuwe pijplijn vertaalt Illumina MiSeq amplicon sequencing-gegevens in hoogwaardige en betrouwbare OTU's, met verbeterde prestaties en nauwkeurigheid.
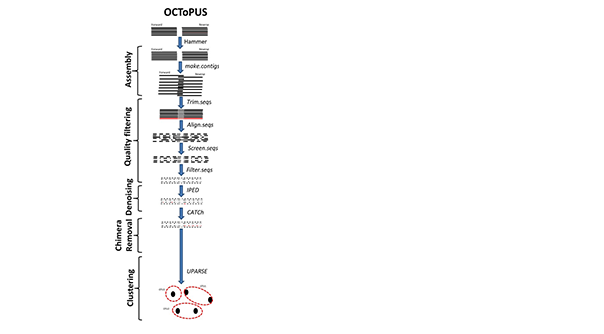
-
Open vacature!
-
Heb je interesse om bij het team te komen?
Als je geïnteresseerd bent om ons team te versterken voor een opleiding of stageproject, neem dan contact met ons op.
- surya [dot] gupta [at] sckcen [dot] be (Dr. Surya Gupta) surya [dot] gupta [at] sckcen [dot] be (📧) 👁️🗨️
- Emre [dot] Etlioglu [at] sckcen [dot] be (Dr. Emre Etlioglu) Emre [dot] Etlioglu [at] sckcen [dot] be (📧)