Bioinformatique
Combiner l'informatique, les statistiques et les mathématiques pour répondre aux questions de la recherche biologique

À propos
Notre équipe de bioinformatique fait partie du groupe d'experts en Biosciences interdisciplinaires (BIO).
Notre recherche est spécialisée dans l'analyse de données omiques, le développement d'algorithmes et de pipelines, l'exploration de données et l'intégration de données multi-omiques. Elle vise à établir des méthodes et des outils de traitement solides, afin de permettre une meilleure compréhension dans le domaine de la protéomique, de la (méta-)génomique et de son intégration avec d'autres données omiques.
Nous fournissons également des solutions bioinformatiques pour un large éventail de données biologiques, à savoir des micro-organismes, des cellules humaines, des souris et des plantes, produites par les laboratoires de microbiologie, de radiobiologie, d'études d'impact sur la biosphère et la recherche sur le cancer du SCK CEN.
Publications récentes
Si vous souhaitez en savoir plus sur vos données ou trouver la solution bioinformatique optimale, nous sommes ouverts à toute collaboration. Pour en savoir plus sur nos travaux de recherche, consultez nos publications récentes.
Expertise du SCK CEN en matière de technologie omique
-

Génomique
- Séquençage du génome entier
- Méta-génomique
- Séquençage des amplicons
-

Épigénomique
- Méthylation du génome entier (réseau, séquençage)
- Méthylation ciblée (réseau, séquençage)
- ChIP-Seq
-

Transcriptomique
- RNAseq sur population de cellules
- Micro RNAseq
- Lnc RNAseq
- RNAseq sur cellules isolées
-

Protéomique
- Spectrométrie de masse
Outils gratuits et open source
Les outils ci-dessous sont des outils gratuits et open source qui sont développés par le SCK CEN. Les travaux de recherche sont accessibles au public afin de soutenir les travaux de recherche ultérieurs de l'ensemble de la communauté des chercheurs.
Outils de synthèse par SCK CEN
-
GenDisCal est destiné à comparer systématiquement un grand nombre de génomes bactériens entre eux. Il peut servir à réunir des génomes inconnus en groupes appartenant à la même espèce, ou comme moyen d'identifier rapidement les espèces auxquelles appartiennent des souches individuelles. GenDisCal est principalement conçu autour de l'utilisation de fréquences d'oligonucléotides courts (3~8bp), mais peut également être utilisé pour calculer l'identité moyenne des nucléotides en utilisant NCBI-Blast (doit être installé séparément) ou une approximation de celle-ci. GenDisCal est également doté d'une interface utilisateur graphique, ce qui le rend facile à utiliser sous Windows et Linux.
-
MAGISTA est une approche sans alignement pour estimer la complétude et la pureté des bins métagénomiques. Pour ce faire, on utilise généralement des gènes marqueurs, mais seuls quelques-uns de ces gènes sont universellement conservés, ce qui limite la précision de ces méthodes pour les nouvelles espèces découvertes par métagénomique. En utilisant des oligonucléotides composés de fragments de 1 kb à 50 kb, MAGISTA tire parti des limites intra-génome sur les distances inter-fragment qui restent cohérentes dans la plupart des espèces bactériennes. En analysant systématiquement les distributions de ces distances inter-fragment à l'aide d'un modèle de forêt aléatoire s'entraînant avec des bins réalistes, MAGISTA peut prédire la complétude et la pureté mieux que les méthodes basées sur les gènes marqueurs qui reposent sur des gènes universellement conservés, bien qu'il soit encore loin d'être précis.
-
BacPipe est un pipeline bioinformatique complet, rapide et à faible coût de calcul pour l'analyse de séquences de génomes entiers bactériens obtenues à partir de technologies de séquençage de deuxième et troisième générations. Les utilisateurs peuvent choisir d'analyser directement les lectures brutes de séquençage ou les contigs ou les scaffolds dans BacPipe. Le pipeline est un ensemble d'outils bioinformatiques de pointe, en accès libre, pour la vérification de la qualité, l'assemblage et l'annotation du génome, et l'identification du génotype bactérien (typage MLST et emm), des gènes de résistance, du ou des plasmides, des gènes de virulence et des polymorphismes mononucléotidiques (SNP). Le module d'épidémie de BacPipe peut être utilisé, avec les SNP et les métadonnées des patients, pour analyser simultanément de nombreuses souches afin de comprendre les relations évolutives et de construire rapidement des voies de transmission bactérienne. Il est important de noter que BacPipe est conçu pour exécuter plusieurs outils simultanément, ce qui réduit considérablement le délai d'obtention des résultats. BacPipe offre un pipeline automatisé qui contribuera à surmonter l'un des principaux obstacles rencontrés par les microbiologistes pour analyser et interpréter les données WGS, facilitant ainsi leur application directe pour les soins de routine aux patients dans les hôpitaux et la surveillance de la santé publique et du contrôle des infections.
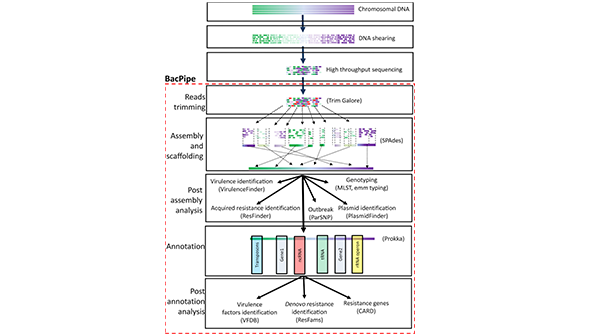
-
L'amplification par PCR de ces gènes marqueurs (16S, 18S, ITS, etc.) produit une quantité importante de séquences artificielles, souvent appelées chimères. CATCh combine deux classificateurs d'apprentissage automatique (basés sur la référence et de novo) développés en intégrant les résultats des outils de détection de chimères existants dans une nouvelle méthode plus puissante. En comparant ces classificateurs avec les outils existants, que ce soit en mode référence ou de novo, une meilleure performance de cette méthode d'ensemble a été observée sur un large éventail de données de séquençage, y compris des ensembles de données simulées, de pyroséquençage 454 et d'Illumina MiSeq. Comme cet algorithme combine les avantages de différents outils individuels de détection des chimères, il produit des résultats plus robustes lorsqu'il est confronté à des séquences chimériques présentant une faible divergence entre les parents, une courte longueur de l'intervalle chimérique et un nombre variable de parents. De plus, il a pu être démontré que l'intégration de CATCh dans le pipeline de prétraitement a un effet bénéfique sur la qualité du clustering dans les unités taxonomiques opérationnelles.
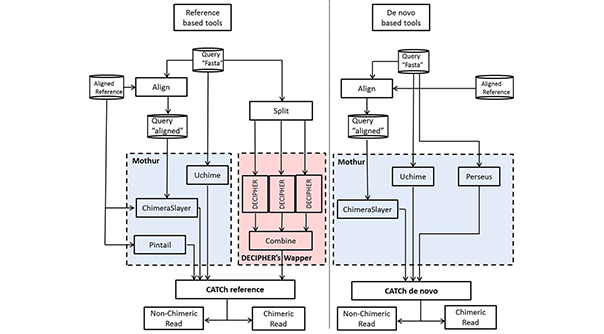
-
Pour les études de métagénomique de l'ARNr 16S, la technologie de pyroséquençage 454 était l'une des plateformes les plus fréquemment utilisées, mais les erreurs de séquençage entraînent toujours d'importants problèmes d'analyse des données (par exemple, dans le regroupement en unités taxonomiques et l'estimation de la biodiversité). NoDe (Noise Detector) est un algorithme de correction d'erreurs conçu pour les données de pyroséquençage 454. Il est entraîné à identifier les positions dans les lectures de séquençage 454 qui sont susceptibles de comporter une erreur, et regroupe ensuite ces lectures sujettes à erreur avec les lectures correctes, ce qui donne une lecture représentative sans erreur. L'effet positif de NoDe dans les études sur l'ARNr 16S a été confirmé par l'effet bénéfique sur la précision du regroupement des lectures de pyroséquençage en unités taxonomiques opérationnelles. NoDe s'est avéré être un algorithme de débruitage efficace sur le plan informatique pour les lectures de pyroséquençage, produisant les taux d'erreur les plus bas dans une étude comparative approfondie avec d'autres algorithmes de débruitage.
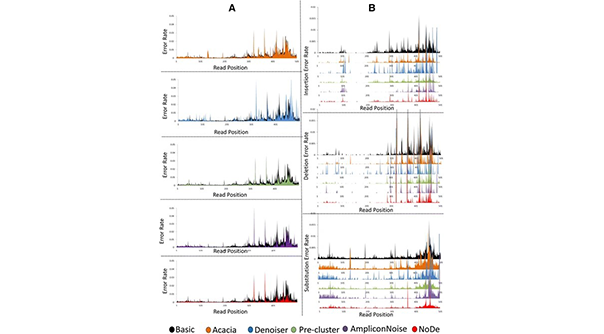
-
IPED est un puissant outil de débruitage permettant de corriger les erreurs de séquençage dans les données de séquençage d'amplicons Illumina MiSeq. Il comprend une méthode d'apprentissage automatique qui prédit les positions potentiellement erronées dans les lectures de séquençage sur la base d'une combinaison de mesures de qualité. Ensuite, ces informations sont utilisées pour regrouper les lectures contenant des erreurs avec les lectures correctes, ce qui permet d'obtenir des lectures consensuelles sans erreur. Ceci est réalisé en masquant les positions potentiellement erronées pendant cette étape de regroupement. La réduction du taux d'erreur a eu un effet positif sur le regroupement des lectures en unités taxonomiques opérationnelles, avec une correspondance presque parfaite entre le nombre de groupes et le nombre théorique d'espèces présentes dans les communautés fictives.
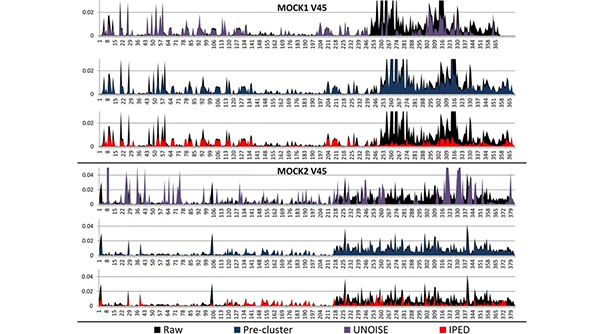
-
DynamiC est un algorithme de regroupement spécifique à une région, basé sur la taxonomie. Il remplace l'utilisation d'un seuil standard (généralement 97 %) pour le regroupement des amplicons 16S en unités taxonomiques opérationnelles (OTU). L'utilisation d'une coupure généralisée est discutable lorsqu'elle est appliquée à des amplicons courts couvrant seulement une ou deux régions variables du gène de l'ARNr 16S. Cela entraînera des biais dans les mesures de diversité et rendra difficile la comparaison des résultats obtenus avec des amplicons dérivés de différents jeux d'amorces. DynamiC, quant à lui, prend en compte les taux d'évolution différentiels des lignées taxonomiques afin de définir un score de coupure de regroupement d'OTU dynamique et dépendant de la taxonomie. Ceci est mis en œuvre en prenant en considération les régions variables amplifiées et la famille taxonomique lors de la définition de ce seuil, un tel seuil conduira à des résultats plus robustes et à une correspondance plus étroite entre les OTU et les espèces.
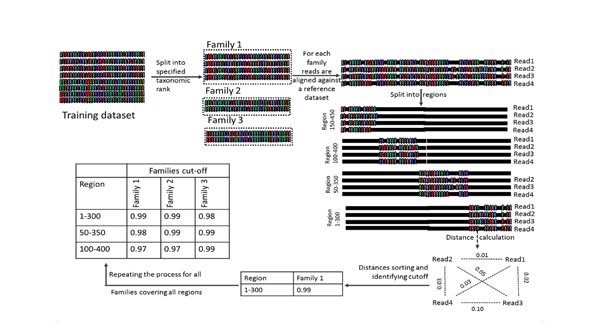
-
OCToPUS est un pipeline complet qui réalise une combinaison optimale de différents algorithmes, dont mothur pour l'alignement et le traitement des lectures, IPED pour le débruitage, CATCh pour la détection des chimères, UPARSE pour le regroupement des OTU et SPAdes pour le filtrage de pré-assemblage. OCToPUS obtient le taux d'erreur le plus faible, le nombre minimal d'OTU parasites et la correspondance la plus étroite avec la communauté existante, tout en conservant le plus grand nombre de lectures par rapport aux autres pipelines (dans l'analyse comparative). Le pipeline nouvellement introduit traduit les données de séquençage d'amplicons Illumina MiSeq en OTU de haute qualité et fiables, avec des performances et une précision améliorées.
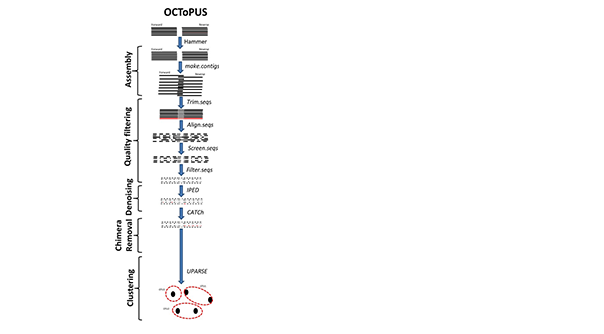
-
Poste vacant !
-
Vous souhaitez rejoindre l'équipe ?
Si vous êtes intéressé et souhaitez rejoindre notre équipe pour un projet de formation ou de stage, vous pouvez nous contacter.
- surya [dot] gupta [at] sckcen [dot] be (Dr. Surya Gupta) surya [dot] gupta [at] sckcen [dot] be (📧) 👁️🗨️
- Emre [dot] Etlioglu [at] sckcen [dot] be (Dr. Emre Etlioglu) Emre [dot] Etlioglu [at] sckcen [dot] be (📧)