Bioinformatics
Combining computer science, statistics & mathematics to address biological research questions

About
Our Bioinformatics team is part of the Interdisciplinary Biosciences Expert Group (BIO).
Our research specialises in omics data analysis, algorithm & pipeline development, data mining and multi-omics data integration with the focus towards establishing solid processing methods and tools, to enable better understanding in the field of proteomics, & (meta-)genomics and their integration with other omics.
We provide bioinformatics solutions for data produced from a wide range of biological system i.e. microorganisms, human cells, mice, and plants, studied at the laboratories of microbiology, radiobiology, biosphere impact studies and cancer research in SCK CEN.
Recent publications
If you like to know more about your data or would like to find the most optimal bioinformatics solution, we are open for collaboration. To know more about our research work, check out our recent publications.
SCK CEN expertise in omics technology
-

Genomics
- Whole genome sequencing
- Meta genomics
- Amplicon sequencing
-

Epigenomics
- Whole genome methylation (array, sequencing)
- Targeted methylation (array, sequencing)
- ChiP-Seq
-

Transcriptomics
- Bulk RNAseq
- Micro RNAseq
- Lnc RNAseq
- Single cell RNAseq
-

Proteomics
-
Shotgun proteomics
-
Metaproteomics
-
Free & open source tools
The tools below developed at SCK CEN are free, open source and have open access publications. These tools allow further support and contribution in the future research work of the scientific community.
Overview tools by SCK CEN
-
GenDisCal is intended to systematically compare large numbers of bacterial genomes against each other. This can be used to cluster unknown genomes into groups belonging to the same species, or as a way to rapidly identify the species to which individual strains belong. GenDisCal is primarily designed around using short (3~8bp) oligonucleotide frequencies, but can also be used to compute average nucleotide identity using NCBI-Blast (must be installed separately) or an approximation thereof. GenDisCal also comes with a graphical user interface, making it easy to use on both Windows and Linux.
-
MAGISTA is an alignment-free approach to estimate completeness and purity of metagenomic bins. Typically this is done using marker genes, but only a few of these genes are universally conserved, which limits the accuracy of such methods for novel species found using metagenomics. By using oligonucleotide composition of 1kb- to 50kb-fragments, MAGISTA leverages intra-genome limits on inter-fragment distances that stay consistent throughout most bacterial species. By systematically analyzing the distributions of these inter-fragment distances using a random forest model training using realistic bins, MAGISTA can predict completeness and purity better than marker-gene-based methods that rely on universally conserved genes, though it is still far from accurate.
-
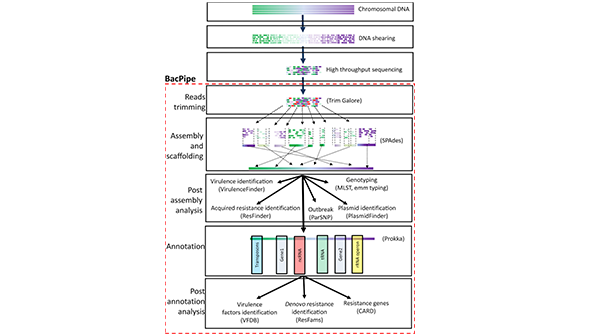
BacPipe is a comprehensive, rapid, and computationally low-resource bioinformatics pipeline for the analysis of bacterial whole genome sequences obtained from second and third-generation sequencing technologies. Users can choose to directly analyse raw sequencing reads or contigs or scaffolds in BacPipe. The pipeline is an ensemble of state-of-the-art, open-access bioinformatics tools for quality verification, genome assembly and Annotation, and identification of the bacterial genotype (MLST and emm typing), resistance genes, plasmid(s), virulence genes, and single nucleotide polymorphisms (SNPs). The outbreak module in BacPipe can be used, along with the SNPs and patient metadata, to simultaneously analyse many strains to understand evolutionary relationships and rapidly construct bacterial transmission routes. Importantly, BacPipe is designed to run multiple tools simultaneously which considerably reduces the time-to-result. BacPipe offer an automated pipeline that will contribute to overcoming one of the primary hurdles faced by microbiologists for analysing and interpreting WGS data, facilitating its direct application for routine patient care in hospitals and public health and infection control monitoring.
-
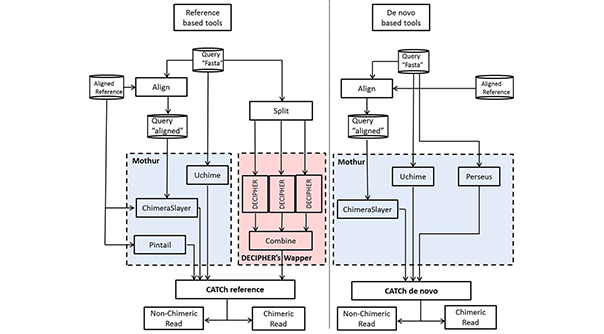
PCR amplification of these marker genes (16S, 18S, ITS, etc) produces a significant amount of artificial sequences, often referred to as chimeras. CATCh combine two machine learning classifiers (reference-based and de novo) developed by integrating the output of existing chimera detection tools into a new, more powerful method. When comparing these classifiers with existing tools in either the reference-based or de novo mode, a higher performance of this ensemble method was observed on a wide range of sequencing data, including simulated, 454 pyrosequencing, and Illumina MiSeq data sets. Since this algorithm combines the advantages of different individual chimera detection tools, it produces more robust results when challenged with chimeric sequences having a low parent divergence, short length of the chimeric range, and various numbers of parents. Additionally, it could be shown that integrating CATCh in the preprocessing pipeline has a beneficial effect on the quality of the clustering in operational taxonomic units.
-
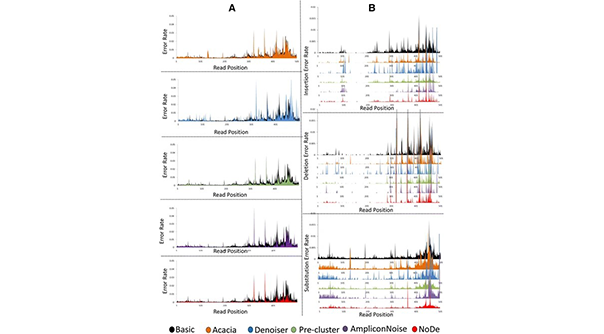
For 16S rRNA metagenomics studies, the 454 pyrosequencing technology was one of the most frequently used platforms, but sequencing errors still lead to important data analysis issues (e.g. in clustering in taxonomic units and biodiversity estimation). NoDe (Noise Detector) is an error correction algorithm designed for 454 Pyrosequencing data. It is trained to identify those positions in 454 sequencing reads that are likely to have an error, and subsequently clusters those error-prone reads with correct reads resulting in error-free representative read. The positive effect of NoDe in 16S rRNA studies was confirmed by the beneficial effect on the precision of the clustering of pyrosequencing reads in operational taxonomic units. NoDe was shown to be a computational efficient denoising algorithm for pyrosequencing reads, producing the lowest error rates in an extensive benchmarking study with other denoising algorithms.
-
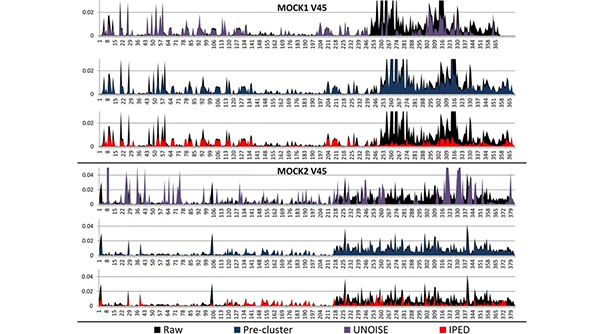
IPED is a powerful denoising tool for correcting sequencing errors in Illumina MiSeq amplicon sequencing data. It includes a machine learning method which predicts potentially erroneous positions in sequencing reads based on a combination of quality metrics. Subsequently, this information is used to group those error-containing reads with correct reads, resulting in error-free consensus reads. This is achieved by masking potentially erroneous positions during this clustering step. Reducing the error rate had a positive effect on the clustering of reads in operational taxonomic units, with an almost perfect correspondence between the number of clusters and the theoretical number of species present in the mock communities.
-
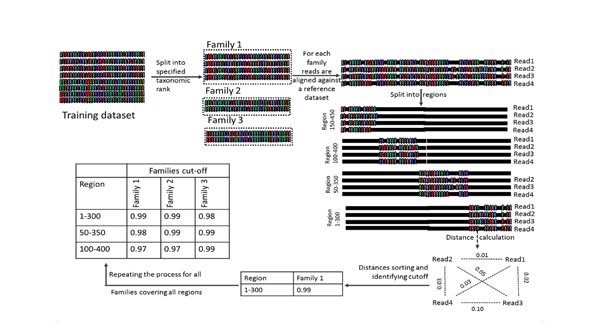
DynamiC is taxonomically driven region-specific clustering algorithm. It replaces the use of a standard cut-off (commonly 97%) for the clustering of 16S amplicons into operational taxonomic units (OTUs). The use of generalised cut-off is questionable when applied to short amplicons only covering one or two variable regions of the 16S rRNA gene. It will lead to biases in diversity metrics and makes it hard to compare results obtained with amplicons derived with different primer sets. DynamiC -on the other hands- takes into account the differential evolutional rates of taxonomic lineages in order to define a dynamic and taxonomic-dependent OTU clustering cut-off score. This is implemented by taking into consideration the amplified variable regions and the taxonomic family when defining this cut-off, such a threshold will lead to more robust results and closer correspondence between OTUs and species.
-
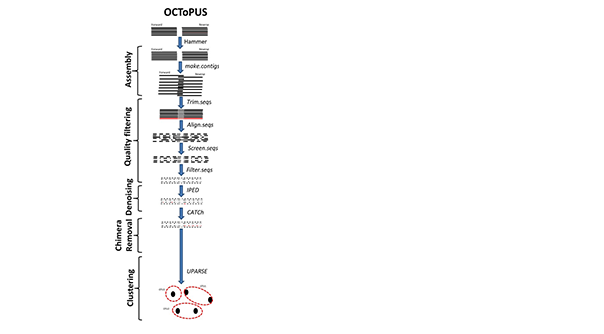
OCToPUS is a comprehensive pipeline that making an optimal combination of different algorithms, which includes mothur: for reads alignment and handling, IPED: for denoising, CATCh: for chimera detection, UPARSE: for OTU clustering and SPAdes for pre-assembly filtering. OCToPUS achieves the lowest error rate, minimum number of spurious OTUs, and the closest correspondence to the existing community, while retaining the uppermost amount of reads when compared to other pipelines (in the comparative analysis). The newly introduced pipeline translates Illumina MiSeq amplicon sequencing data into high-quality and reliable OTUs, with improved performance and accuracy.
-
Current opening
-
Are you interested to join the team?
If you are interested to join Bioinformatics team for training or internship project, you can contact:
surya [dot] gupta [at] sckcen [dot] be (Dr. Surya Gupta) surya [dot] gupta [at] sckcen [dot] be (📧) 👁️🗨️
Emre [dot] Etlioglu [at] sckcen [dot] be (Dr. Emre Etlioglu) Emre [dot] Etlioglu [at] sckcen [dot] be (📧)